Эпигенетические нарушения при гемобластозах
- Вступление
- Что такое эпигенетика?
- Структура хроматина
- Метилирование ДНК
- Модификации гистонов
- РНК-ассоциированное подавление экспрессии генов
- Заключение
- Cписок литературы
Вступление
Изучение хромосомных аберраций, ведущих к повреждению нормальной экспрессии генов при гемобластозах, позволило выявить, что в опухолевой клетке, кроме нарушения структуры ДНК, изменения обнаруживаются на эпигенетическом уровне, т.е. на уровне считывания генетической информации с участием белковых и нуклеотидных структур. При этом если значение генетических аберраций при этих заболеваниях в настоящее время достаточно хорошо изучено, эпигенетические нарушения, ассоциированные с развитием лейкемического фенотипа, стали предметом пристального внимания лишь в последнее время.
Что такое эпигенетика?
По современному определению, к эпигенетически обусловленным относят все унаследованные клеткой в процессе деления (митоза или мейоза) особенности регуляции экспрессии генов, не связанные непосредственно с изменением кода ДНК [1]. Осуществляют эпигенетическое влияние на генную экспрессию процессы, влияющие на степень конденсации хроматина – структуры-носителя генетической информации в клетке, состоящей из ДНК и особых белков-гистонов.
При уменьшении конденсации хроматина освобождается доступ факторов транскрипции к ДНК и становится возможным экспрессия генов, то есть последовательное образование РНК (транскрипция) и белка (трансляция). Ведущими процессами, регулирующими структуру хроматина, являются метилирование ДНК, ферментная модификация белков-гистонов и РНК-асссоциированное подавление транскрипции и трансляции [2]. Исследования последнего времени выявили устойчивую связь между всеми тремя компонентами эпигенетической регуляции, их способность к взаимной активации (рис. 1).
Оказалось, что нарушения в одной из систем неизбежно приводит к повреждению механизмов двух других, и, таким образом, вызывает неадекватную экспрессию или, наоборот подавление функционирования огромного количества генов. В связи с обнаружением различных видов нарушений этих процессов, появилась новая концепция развития целого ряда заболеваний – эпигенетическая, возникли термины «эпигенетические болезни» и «эпигенетическая терапия».
Рис. 1. Взаимосвязь механизмов эпигенетической регуляции.

Структура хроматина
Для лучшего понимания процессов считывания генетической информации и их регуляции следует вспомнить, что генетический материал в клетке существует в виде хроматина – структуры, состоящей из ДНК и белков – гистонов. Структурной единицей хроматина является нуклеосома, в состав которой входят часть спирали ДНК, размером 146 пар нуклеотидов (п.н.), совершающая виток в 1,7 раз вокруг белковой структуры – октамера, состоящей из гистонов.
Нуклеосомы связаны между собой участком ДНК около 50 п.н. [3]. Таким образом, хроматин образует динамическую структуру, позволяющую компактно расположить в ядре клетки около 2 м ДНК, сохранив при этом ее способность к функционированию. Для осуществления процесса транскрипции – образования РНК, белкам-транскрипционным факторам необходим доступ к регуляторным последовательностям ДНК. Доступ регулируется двумя взаимосвязанными процессами: метилированием ДНК и ацетилированием (и другими ферментными модификациями) гистонов, которые будут рассмотрены ниже. Схематически процесс изображен на рис. 2.
Рис. 2. Изменение структуры хроматина в процессе метилирования ДНК и ацетилирования гистонов

Метилирование ДНК
Метилированием называется присоединение группы СН3 к атому углерода в 5 позиции азотистого основания цитозина, одного из четырех нуклеотидов, составляющих структуру цепи ДНК (рис. 3). Процесс метилирования является одним и фундаментальных процессов эпигенетического подавления экспрессии генов, закрепленным в процессе эволюции [4,5]. Образование метилцитозина ведет к конденсации хроматина, препятствует присоединению транскрипционных факторов к своим мишеням на ДНК и, таким образом, ингибирует считывание информации.
В процессе естественного развития метилирование является ведущим механизмом осуществления последовательной ткане-специфической экспрессии генов и правильного развития эмбриона, инактивации одной Х-хромосомы у женщин и осуществления механизма импринтинга, позволяющего «замалчивать» один из родительских генов в диплоидном наборе хромосом.
Метилированию в норме подвергается от 2 до 7% всех цитозиновых остатков ДНК клетки. При этом в 70% случаев цитозин метилируется в составе динуклеотидов С-G (CpG). СpG участки, как правило, представляют собой фрагменты ДНК длиной более чем 500 пар нуклеотидов (п.н.) и базируются в зонах инициации считывания информации – промотерах – более чем 40% генов млекопитающих [6,7].
Осуществляется метилирование с помощью ферментов ДНК-метилтрансфераз (DNMT). Метилтранферазы можно условно разделить на две группы – метилирующие ДНК de novo, то есть в тех участках, где ранее не было метилцитозина (DNMT3A и DNMT3B), и «поддерживающие» метилирование в дочерней цепи ДНК, образующейся в процессе репликации (DNMT1), сохраняя, таким образом, структуру, присущую материнской цепи.
Рис. 3. Процесс метилирования

Нарушения метилирования при развитии злокачественных новообразований возникают, как правило, на ранних стадиях. Общий низкий уровень метилирования цитозина (гипометилирование) при этом сочетается с обратным процессом -гиперметилированием CpG участков в промотерах генов-супрессоров опухоли [8,9]. Тотальное гипометилирование ведет к повышению экспрессии протоонкогенов, генов, кодирующих ростовые факторы, а также целого ряда генов, таких, как ген активатора плазминогена урокиназного типа (PLAU), ген гепараназы и кальций-связывающего протеина (S100A4), способствующих метастазированию путем проникновения клетки через стенки кровеносных и лимфатических сосудов [10].
Важную роль играет также гипометилирование специфических олигонуклеотидных элементов – ретротранспозонов – имеющих вирусное происхождение. Ретротранспозоны представляют собой обратные транскрипты вирусной РНК, в норме достаточно произвольно встроенные в структуру хозяйской ДНК. Обычно эти элементы метилированы и поэтому неактивны. Зачастую ретротранспозоны встраиваются в регуляторные зоны генов – интроны. Активация этих элементов при опухолевой трансформации ведет к повышению генетической нестабильности и нарушению нормальной экспрессии хозяйских генов [11].
Причины гипометилирования ДНК в злокачественных клетках до конца не ясны. Гипотеза, предполагающая причиной общий или частичный дефицит метилирующих ферментов не объясняет одновременное гиперметилирование промотеров целого ряда генов – супрессоров опухоли. Некоторые исследователи предполагают, что причиной тотального гипометилирования ДНК в злокачественных клетках является образование определенных изоформ фермента ДНК-метилтрансферазы типа DNMT3B.
Эта форма фермента, связываясь с CpG участками промотеров генов, не приводит к их метилированию, но при этом препятствует связи этих участков с активными формами метилтрансфераз [12,13]. В то же время этот тип фермента и, в меньшей степени, DNMT1 способствуют активному метилированию и подавлению экспрессии генов-супрессоров опухоли. При ОМЛ характерным является высокий уровень экспрессии как DNMT3B, так и DNMT1, что сопровождается гиперметилированием промотеров таких генов – опухолевых супрессоров, как ингибиторы циклин-зависимой киназы (CDKN2A и CDKN2B), ген эстрогенового рецептора I (ESR1), и ген ретинобластомы (RB1) [14]. В последние годы появились данные целого ряда исследований, доказывающих, что ДНК-метилтрансферазы участвуют в образовании белковых комплексов, подавляющих транскрипцию, которые возникают при участии химерных протеинов, таких, как PML/RARA и AML/ETO (1, 15, 16, 17).
Открытие роли метилирования в онкогенезе привело к широкому внедрению препаратов-ингибиторов ДНК-метилтрансфераз. Наиболее известным из них является 5-аза-2’-деоксицитидин (Дакоген), который, встраиваясь в цепочку ДНК вместо цитозина, образует прочную ковалентную связь с ДНК-метилтрансферазами, препятствуя, таким образом, метилированию и снимая блок с репрессированного ранее гена [18].
Модификации гистонов
Следующим важнейшим механизмом эпигенетической регуляции является ферментная модификация основных белков хроматина – гистонов. Гистоны образуют ядро нуклеосомы, вокруг которого формируется виток спирали ДНК. Существует 4 вида белков-гистонов. При формировании ядра нуклеосомы они образуют следующие тесно связанные структуры: тетрамер из протеинов Н3 и Н4 и 2 димера из Н2А и Н2В [19]. Схематически ядро нуклеосомы представлено на рис. 4. Гистон Н1 у высших эукариот осуществляет связывание нуклеосом между собой, способствуя конденсации хроматина [20].
Рис. 4. Модель ядра нуклеосомы (вид сверху): тетрамер Н3 и Н4 и один из димеров Н2А и Н2В (второй димер расположен под первым)

Структура каждого гистона включает глобулярное ядро, обладающее специфическим мотивом – завиток-петля-завиток, осуществляющим димеризацию и свободный «хвостовой» домен, включающий аминокислоты лизин, аргинин и серин. Этот домен обладает положительным электрическим зарядом, и, взаимодействуя с анионными группами цепи ДНК определяет стабильность структуры нуклеосомы [21].
Ацетилирование или метилирование свободной аминогруппы лизина приводит к изменению общего заряда белка и, соответственно, изменению структуры хроматина – освобождается доступ транскрипционных факторов к ДНК (см. рис. 3). Такие же изменения происходят при фосфорилировании гидроксильного остатка серина. В целом, в настоящее время известно более 50 позиций в аминокислотной структуре гистонов, которые могут быть подвержены ферментной модификации (метилированию, ацетилированию, фосфорилированию, АДФ-рибозилированию и т.д.) [22].
Оказалось, что каждое из этих изменений является своеобразным знаком для привлечения различных регуляторных комплексов, каждый из которых определяет особенности транскрипции. Таким образом, сочетание различных модификационных процессов создает так называемый «гистоновый код», во многом определяющий сущность и порядок считывания генетической информации. Оказалось, что информационные возможности гистонового кода едва ли не больше возможностей классического кода ДНК [23, 24].
Процессы ацетилирования и деацетилирования, наиболее изученные к настоящему времени, осуществляются специфическими группами ферментов – гистонацетилазами (или ацетилтрансферазами), или HAT и гистон-деацетилазами, или HDAC. Гистонацетилазы работают, как правило, в составе больших комплексов, которые, зачастую, способны изменять их активность. Интересно, что целый ряд известных транскрипционных факторов таких, как CBP и p300, TAFII250, SRC-1 имеют последовательности, гомологичные структуре HAT, и обладают способностью к самостоятельному ацетилированию гистонов.
Антагонисты ацетилаз – деацетилазы составляют 3 класса, отличающиеся по своей структуре и несколько различающиеся по функции. Классической функцией HDAC является репрессия транскрипции с помощью удаления ацетиловой группы с лизиновых остатков и, соответственно, конденсации структуры нуклеосомы [25]. Однако в последнее время появились данные, что в ряде случаев деацетилирование необходимо для успешного функционирования транскрипционно активных генов [17]. Эти данные подтверждают необычайную сложность и, зачастую, парадоксальность гистонового кода.
Участие гистондеацетилаз в лейкемогенезе хорошо изучено на классических примерах острого промиелоцитарного лейкоза (ОПЛ) и ОМЛ, несущих транслокацию t(8;21) или сопровождающихся аномалиями 16 хромосомы.
При ОПЛ в 95% случаев образуется транслокация t(15;17), ведущая к появлению химерного белка PML/RARA. В норме продукт одного из генов-участников транслокации ядерный рецептор RARA образует комплекс с корепрессорами (SMRT, N-CoR, Sin3a) и гистондеацетилазой HDAC, блокирующий транскрипцию регулируемых генов. Этот комплекс разрушается при присоединении естественного лиганда RARA – ретиноевой кислоты – и транскрипция становится возможна.
При образовании химерного протеина PML/RARA репрессирующий комплекс не реагирует на ретиноевую кислоту в физиологических концентрациях. Осуществляемое гистондеацетилазой HDAC деацетилирование гистонов служит также сигналом для активации других ферментов: гистоновых метилтрансфераз и ДНК-метилтрансфераз, также включающихся в репрессирующий комплекс (рис. 5).
Таким образом осуществляется взаимодействие между двумя механизмами эпигенетической регуляции: модификацией структуры гистонов и метилированием ДНК. Фармакологические концентрации ретиноевой кислоты, в тысячи раз превышающие физиологические, способны вызвать диссоциацию репрессирующего комплекса и запустить механизм дифференцировки [1]. Одновременно ретиноевая кислота приводит к активации «лиганда смерти» – TRAIL (TNF-related apoptosis-inducing ligand), являющегося ключевым компонентом механизма апоптоза [26].
Полностью трансретиноевая кислота (АТРА), как «дифференцирующий агент» в терапии ОПЛ представляет собой типичный пример препарата с эпигенетическим действием: не изменяя структуру ДНК, она приводит к разрушению репрессирующего комплекса, деацетилирующего гистоны регуляторных элементов генов, ответственных за дифференцировку клеток.
Тем не менее, несмотря на достигнутые успехи в понимании механизма лейкемогенеза при ОПЛ остается ряд вопросов, требующих дальнейших исследований: 1) взаимодействует ли химерный онкоген при действии АТРА абсолютно с тем же активирующим комплексом, что и нормальный ядерный рецептор; 2) какова роль доменов, принадлежащих гену PML; 3) значение экспрессии реципрокного химерного протеина и т.д.
Рис 5. Процесс нормального функционирования ядерного рецептора RARA и ингибирование транскрипции химерным протеином PML/RARA

В незначительном числе случаев ОПЛ, сопровождающихся образованием транслокации t(11;17) (PLZF/RARA) и t(11;17) STAT5b/RARA, химерные протеины образуют более прочную связь с репрессирующим комплексом за счет дополнительных сайтов в структуре второго участника химерного белка – продукта генов PLZF или STAT5.
Эта связь не разрушается в присутствии высоких концентраций ретиноевой кислоты именно за счет дополнительных связей репрессирующего комплекса. Следует отметить, что в эксперименте in vitro комбинация полностью трансретиноевой кислоты и ингибиторов гистондеацетилаз, разрушающих дополнительные связи химерных протеинов, позволяет добиться дифференцировки лейкемических клеток [27].
При ОМЛ, сопровождающихся t(8;21) или аномалиями 16 хромосомы с вовлечением генов ЕТО(MTG8), AML1(RUNX1) и CBFβ, происходит нарушение образования нормального транскрипционного комплекса CBF. Этот комплекс состоит из продуктов генов c/EBPα, AML1(RUNX1) и CBFβ и обладает гистонацетилазной активностью, потенцируя экспрессию большой группы генов, отвечающих за нормальную дифференцировку гемопоэтических клеток.
При возникновении генетических аберраций, изменяющих нормальную структуру любого из белков комплекса CBF, нарушается его связь с фактором-гисонацетилазой р300, коактиватором и ядерным рецептором TIF2. При транслокации t(8;21) ведущую роль в нарушении комплекса играет та часть химерного протеина AML/ETO, которая считывается со второго участника транслокации – гена ЕТО(MTG8). В норме этот ген экспрессирует белок, участвующий в репрессирующем комплексе и связывающий уже известные нам гистондеацетилазу HDAC1 и корепрессоры N-CoR и Sin3a.
За счет доменов белка ЕТО, осуществляющих эти связи, химерный протеин AML/ETO приобретает функции репрессора транскрипции и подавляет гены в норме активирующиеся AML1 [28]. В последнее время исследуется огромное количество химических соединений, обладающих функцией ингибиторов гистондеацетилаз, однако большинство из них пока проходят первую или вторую фазу клинических испытаний [29].
Весьма сложными нарушениями сопровождаются повреждения нормального функционирования гена MLL, кодирующего протеин, который обладает в том числе и гистон-метилтрансферазной активностью. Основной функцией этого белка является участие в огромном транскрипционном комплексе, в состав которого входят по меньшей мере 29 других белков, вовлеченных как в ацетилирование, так и в деацетилирование гистонов, в метилирование гистонов и ДНК [30].
Этот комплекс осуществляет регуляцию транскрипции генов гомеобокса, участвующих в эмбриональном развитии и экспрессирующихся в ранних предшественниках гемопоэза. В различных транслокациях с участием MLL (которых известно около 60 для всех видов лейкемии) образуются химерные протеины, потерявшие метилтрансферазную активность за счет отсутствия соответствующего домена в составе химерных генов. Скорее всего, механизм лейкемогенеза связан в данной ситуации с нарушением функционального баланса образующегося транскрипционного комплекса, а также, несомненно, от функций, привнесенных вторым участником транслокации [16].
РНК-ассоциированное подавление экспрессии генов
Роль РНК в подавлении экспрессии генов в последнее время привлекает все большее внимание исследователей. Оказалось, что РНК-индуцированная ингибиция может осуществляться на различных этапах считывания генетической информации. В основе этого процесса лежит феномен интерференции – способность двухцепочечной РНК к эффективному ингибированию экспрессии генов. Двухцепочечная молекула РНК расщепляется на фрагменты длиной 21-25 нуклеотидов, образуя так называемую siRNA (small interfering RNA) [31].
Такие молекулы РНК способны образовывать комплексы с протеинами (RISC – RNA induced silencing complex) и вызывать деградацию матричной РНК, то есть останавливать генную экспрессию на посттранскрипционном уровне – не допускать синтеза белка после успешного образования РНК [32]. Ранее этот механизм считался более характерным для клеток растений, однако в последнее время появились данные, что посттранскрипционная ингибиция возникает и в клетках млекопитающих [31].
РНК участвует также и в подавлении экспрессии генов на уровне транскрипции, вызывая РНК-зависимое метилирование ДНК. Образующиеся из двухцепочечной РНК короткие фрагменты имеют структуру, гомологичную промотерам ряда генов и способны индуцировать метилирование этих промотеров, присоединяясь к ним и активируя ДНК-метилтрансферазы [33].
Более того, siRNA способны вызывать метилирование гистонов, подтверждая тем самым взаимосвязь механизмов эпигенетической регуляции между собой, хотя пока до конца не ясен порядок этих событий и их взаимозависимость [34]. Вполне возможно, что возникающее первоначально метилирование лизина в структуре гистона Н3 ведет в дальнейшем к активации ДНК-метилтрансфераз [35]. Не совсем ясен пока и механизм доставки siRNA к геномной ДНК.
В настоящее время рассматривается несколько возможных вариантов участия siRNA в ингибировании экспрессии генов. Первый механизм – прямое связывание РНК со специфическим транспортным белком Argonaute 2 (возможно в комплексе с гистондеацетилазами) и непосредственное связывание структуры с комплементарной последовательностью в промотере гена с дальнейшим включением механизма деацетилтрования гистонов и остановкой транскрипции.
Два других механизма предполагают связывание siRNA с известными репрессирующими комплексами – Mi2/NuRD и Sin3. Первый из них включает гистондеацетилазы (HDAC 1, 2) и ДНК-метилтрансферазу (DNMT1), т.е. обладает способностью к метилированию ДНК и деацетилированию гистонов. Второй имеет в своем составе гистондеацетилазы (HDAC 1, 2) [35].
Схематически все три механизма изображены на рис. 6.
Рис. 6. Возможные механизмы РНК-ассоциированной ингибиции экспрессии генов.
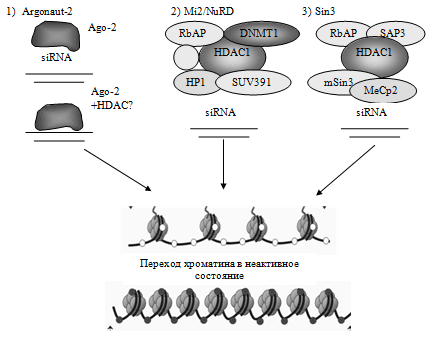
Следует отметить, что пока нет достоверных данных о роли РНК-опосредованного механизма ингибиции генной экспрессии непосредственно в лейкемогенезе, однако исследование этого феномена будет иметь огромное значение для понимания взаимосвязи различных видов эпигенетической регуляции между собой.
Заключение
Изучение эпигенетической регуляции в последние годы позволило не только добиться выдающихся успехов в понимании механизмов канцерогенеза в целом и лейкемогенеза в частности, но и открыть новую область в терапии – эпигенетическую терапию злокачественных заболеваний. Если генетические нарушения не могут быть скорректированы с помощью влияния извне, и ведут к необратимой потере функции поврежденного гена, нарушения эпигенетической регуляции потенциально обратимы, что позволяет рассчитывать на возможность коррекции имеющихся дефектов с помощью правильно подобранного терапевтического воздействия.
Применение так называемой направленной терапии, веществ, модифицирующих действие гистондеацетилаз и ДНК-метилтрансфераз, синтетических siRNA сделает возможным изменение цикла жизни злокачественной клетки, возврат ее к нормальной дифференцировке и естественному апоптозу, что, несомненно, приведет к возникновению качественно нового этапа в лечении онкологических заболеваний.
Литература:
- Altucci L., Clarke N., Nebbioso A et al. Acute myeloid leukemia: Theraputic impact of epigenetic drugs Int Journ of Biochem and Cell Biol, 2005, 37, 1752-1762
- Egger G., Liang G, Aparicio A., Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature, 2004, 429, 457-463
- Kornberg RD, Lorch U. Twenty-five years of nucleosome, fundamental particle of the eukaryote chromosome. Cell, 1999, 98, 285-294
- Holiday R, Pugh JE. DNA modification mechanism and gene activity during development. Science, 1975, 187, 226-232
- Riggs AD. X inactivation, differentiation and DNA methylation. Cytogenet Cell Genet, 1975, 14, 9-25
- Takai D, Jhones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci USA , 2002, 99, 3740-3745
- Monk M. Epigenetic programming of different gene expression in development and evolution. Dev Genet 1995, 17, 188-197
- Baylin SB, Esteller M, Rountree MR et al. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer Hum Mol Genet 2001, 10, 687-692
- Laird PW. Cancer epigenetic. Hum Mol Genet 2005, 14, 65-76
- Luczak MW, Jagodzinski PP. The role of DNA methylation in cancer development. Fol Hist et Cytogen 2006, 44, 143-154
- Carnell AN, Goodman JI. The long (LINEs) and short (SINEs) of it: altered methylation as a precursor of toxicity. Toxicol Sci 2003, 75, 229-235
- Saito Y, Kanai Y, Sakamoto M et al. Expression of mRNA of DNA methyltransferases and methyl-GpC-binding proteins and DNA methylation status on GpC islands and pericentromeric satellite regions during human hepatocarcinogenesis. Hepatology, 200, 33, 561-568
- Weisenberger DJ, Velicescu M, Cheng JC et al. Role of DNA-methyltransferase variant DNMT3b3 in DNA methylation. Mol Cancer Res 2004, 2, 62-67
- Melki JR, Vincent PC, Clark SJ. Concurent DNA hypermethylation of multiple genes in acute myeloid leukemia. Cancer Res 1999, 59, 3730-3740
- Liu S, Shen T, Huynh L. Interplay of RUNX1/MTG8 and DNA methyltransferase 1in acute myeloid leukemia. Cancer Res 2005, 65, 1277-1284
- Di Croce L. Chromatin modifying activity of leukemia associated fusion proteins. Human Mol Genet 2005, 14, 77-84
- Ducasse M, Brown M. Epigenetic aberrations and cancer. Molecular Cancer 2006, 5, 60-70
- Christmas JK. 5-Azacytidine and 5-aza-2-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483-5495
- Luger K, Madeer AW, Richmond RK et al. Crystal structure of the nucleosome core particle at 2.8 resolution. Nature 1997, 389, 251-260
- Francis NJ, Kingdton RE, Woodcock CL. Chromatin compaction by a Polycomb group protein complex. Science 2004, 306, 1574-1577
- Hayes JJ, Clark DJ, Wolffe AP. Histone contributions to the structure of DNA in the nucleosome. Proc Natl Acad Sci USA 1991, 88, 6829-6833
- Jenuwein T. The epigenetic magic of histone lysine methylation. Febs J 2006, 273, 3121-3135
- Jenuwein T., Allis CD. Translating the histone code. Science 2001, 293, 1074-1080
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature 2000, 403, 41-45
- Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol 2004, 338, 17-31
- Clarke N, Nebbioso A, Altucci L, Gronmeyer H. TRAIL. At the center of drugable anti-tumor pathways. Cell Cycle 205, 4, 914-918
- Lin RG, Nagy L, Inoue S. et al. Role of histone deacetylase complex in acute promyelocytic leukemia. Nature 1991, 391, 811-814
- Hiebert SW, Reed-Inderbitzin EF, Amann J et al. The t(8;21) fusion rotein contacts co-repressors and histone deacetylases to repress the transcription of the p14ARF tumor repressor. Blood Cells Mol Dis 2003, 30, 177-183
- Marks PA, Miller T, Richon VM. Histone deacetylases. Curr Opin Pharmacol 2003, 3, 344-351
- Nakamura T., Mori T, Tada S et al. ALL-1 s a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulations. Mol Cell, 10, 1119-1128
- Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes and Development 2001, 15, 188-200
- Zeng Y, Cullen BR. RNA interference in human cells is restricted to the cytoplasm. RNA 2002, 8, 855-860
- Morris KV. Small interference RNA-induced transcriptional gene silencing in human cells. Science 2004
- Kawasaki H, Taira K. Induction of DNA methylation and gene silencing in human cells. Nature 2004
- Kawasaki H, Taira K, Morris KV. siRNA induced transcriptional gene silencing in mammalian cells. Cell Cycle 2005, 4, 442-448